|
How to interpret results?
Let's get an example using the GTPase HRas protein (HRAS, UniprotID P01112)
Insert your job id on the top-right textbox of the screen, and ask for the results: 
The top part of the results will display general information about your target protein: 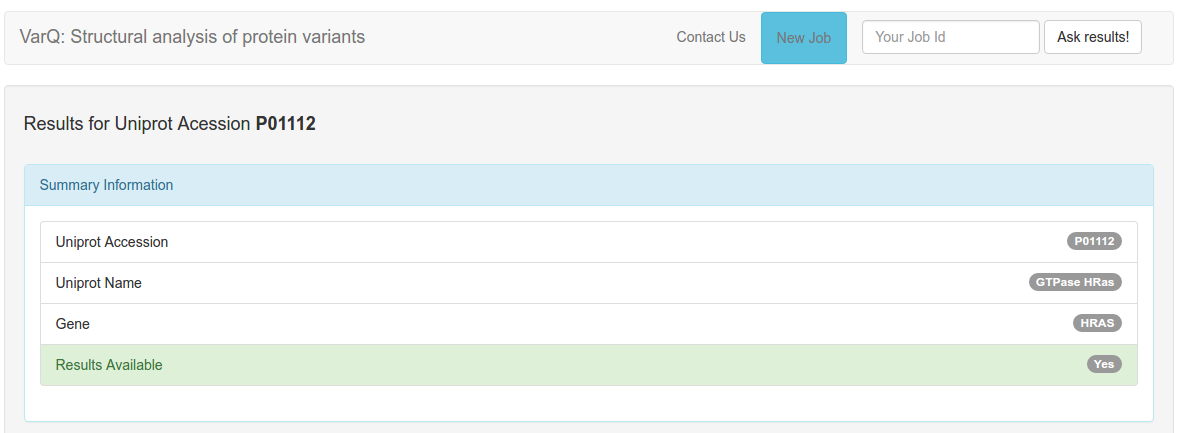
Behind, for each analyzed crystal (press on the help button or read above to know which crysals are taken into account) detailed information is displayed: 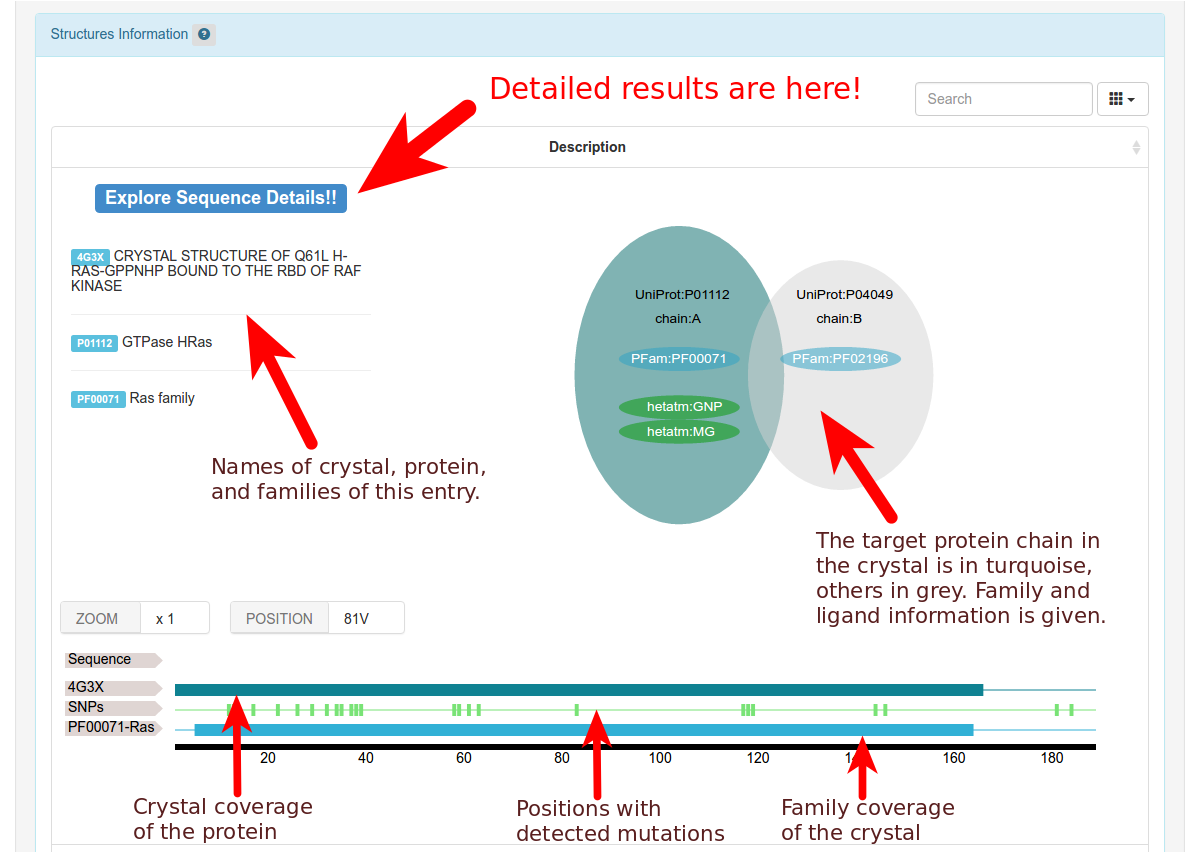
Clicking on any help button will display a modal dialog with detailed information: 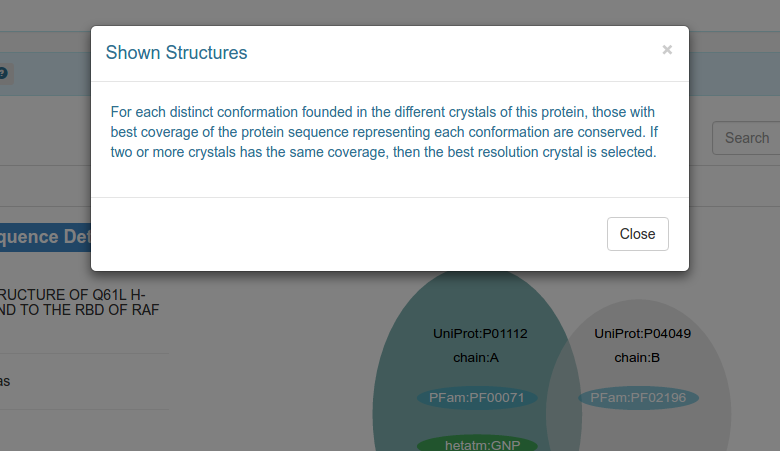
If you click to explore the sequence detail, general information of each position will be listed: 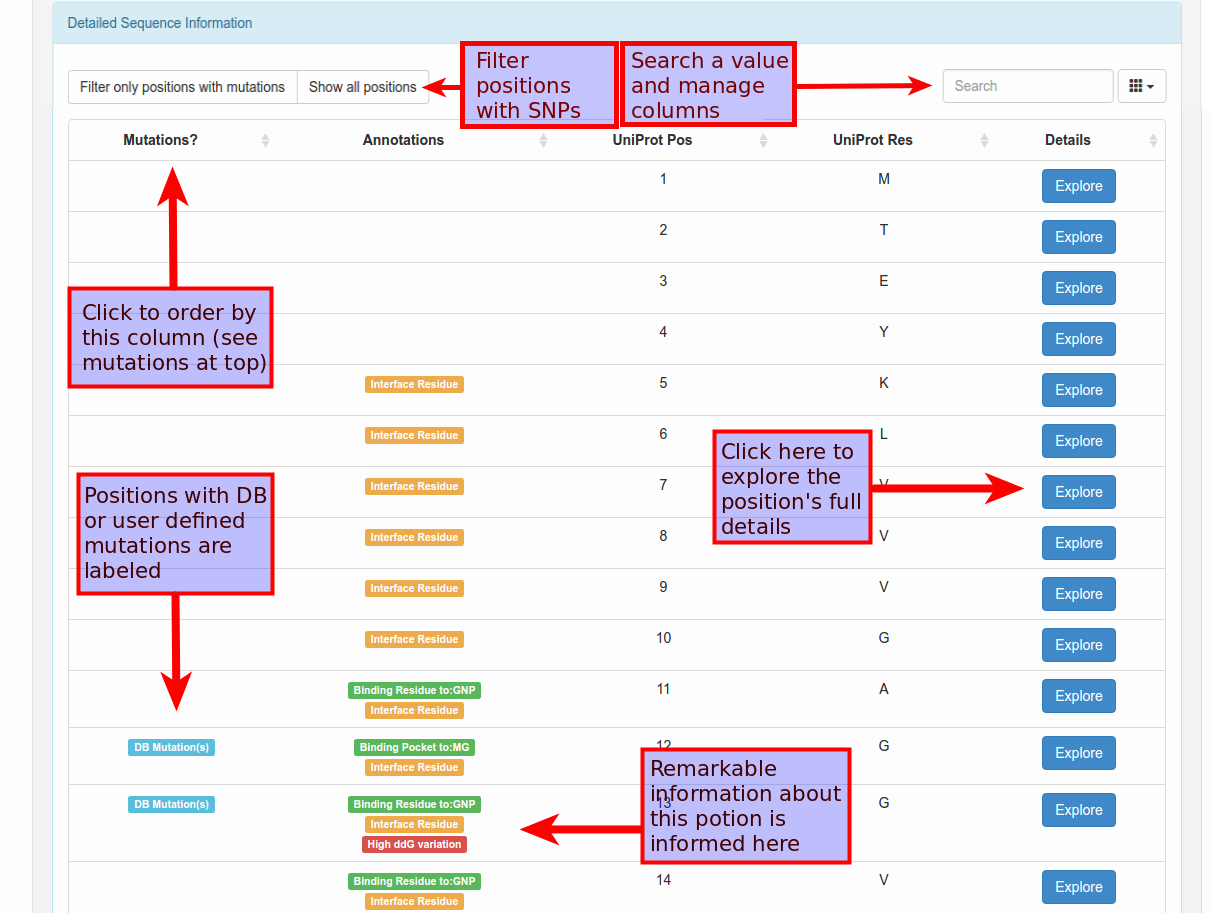
And clicking to explore the detais of the position, all the information about the position will be displayed: 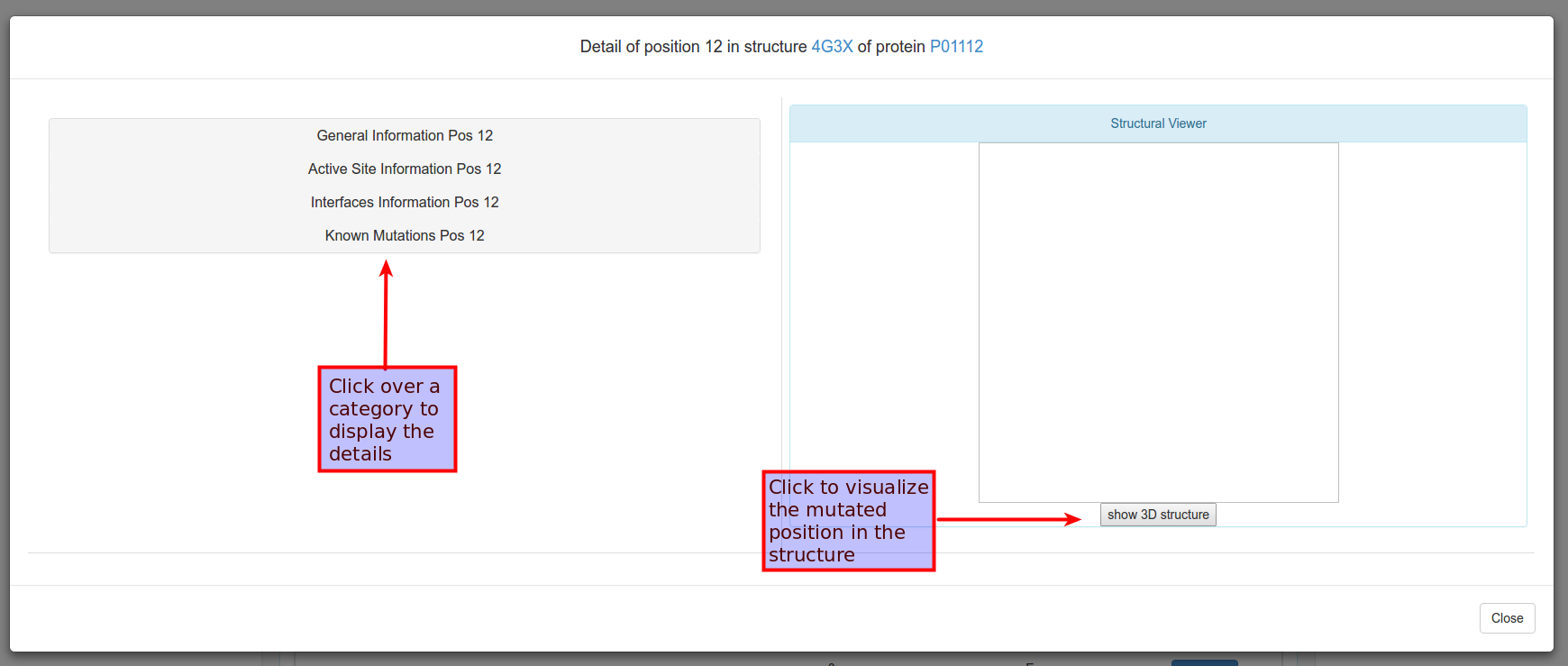
The general information panel contains information about general properties of the specific position. The bFactor is compared in the crystal against all the residues to say how rigid or mobile is relative to the whole experiment. 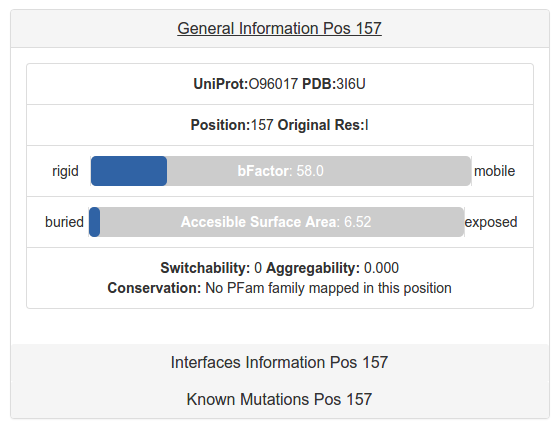
The Interfaces Information tab presents to the user two different aspects of how the selected residue could belong or not to an interface: if the residue have at least one atom at 5 Armostrong or less of any atom of other chain the residue is labeled as "interface residue" (see help). Also, the 3did database could bring to the user valuable information about the observation of this position in his PFam family interfacing with other families in other crystals(see help). 
The Active Site information tab presents, if applies, information about if the residue is marked in the structure as in contact to any ligand or ion, or if it is in the same pocket of a residue that is in contact. This information could be useful to interpret if a mutation could or not affect the protein function. 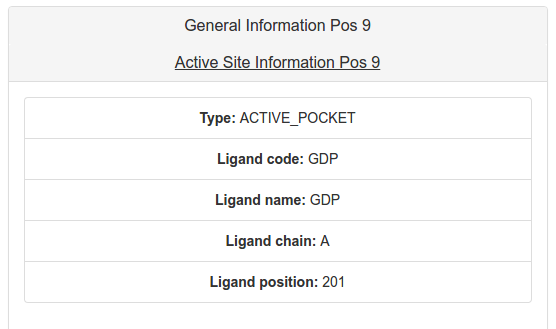
The Known Mutations tab presents, if applies, information about each mutation reported for the position that is been explored. A general description obtained from the reporting database and the variation energy that provokes in the structure is presented. Detailed information could be obtained for deseases information following the link to swissvar database. 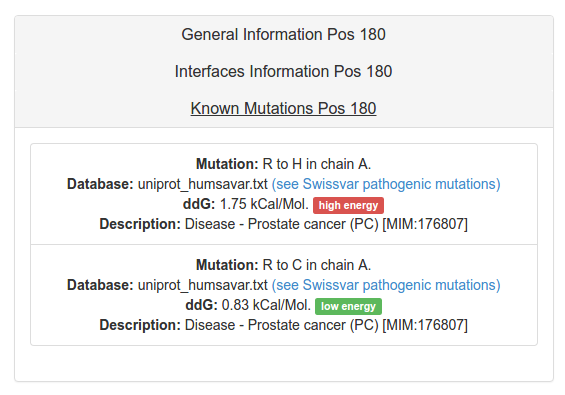
The Structural Viewer tab presents a JMol visualizer to observe the position that the residue explored occupies in the selected crystal. Also, all the heteroatoms are shown to evaluate if a mutation in the current position could affect the protein function or dont. The Jmol tool allows to modify the visualization in several ways and the user has the posibility to do it in this screen. 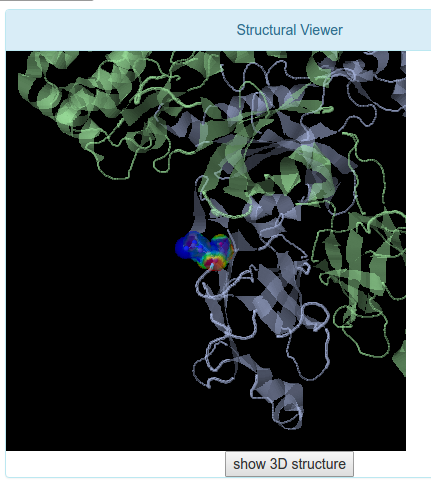
|
Tutorials and examples
Tutorials:
Tutorial: Job loading instructions. Tutorial: Explained output. Help: Interpreting results. Sample results: PAH: Phenylalanine-4-hydroxylase NRAS: GTPase NRas HRAS: GTPase HRas KRAS: GTPase KRas BRAF: Serine/threonine-protein kinase B-raf RAF1: proto-oncogene serine/threonine-protein kinase RASA1: Ras GTPase-activating protein 1 NF1: Neurofibromin MAP2K1: Dual specificity mitogen-activated protein kinase kinase 1 MAP2K2: Dual specificity mitogen-activated protein kinase kinase 2 PTPN11: Tyrosine-protein phosphatase non-receptor type 11 SOS1: Son of sevenless homolog 1 SPRED1: Sprouty-related, EVH1 domain-containing protein 1 |