|
How to interpret results?
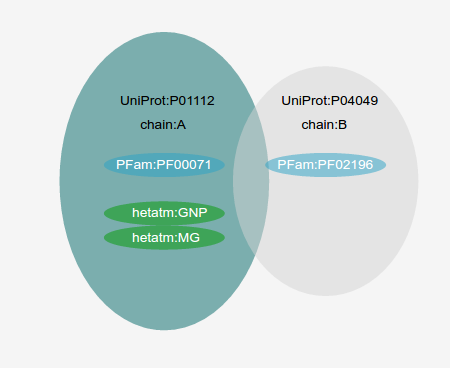
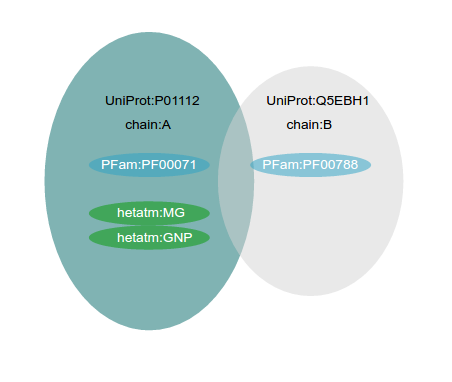
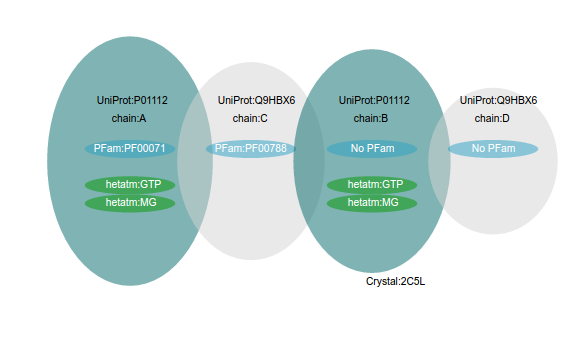
Structure MiningWhen the user specify an UniProt Accession to be analyzed, the VarQ Pipeline searches within all the available structures those that cover different segments of the sequence, preferring those with more coverage first and those with best resolution in second place as tiebreaker. Only those crystals covering at least 20 aminoacids of the sequence of the target protein are considered. If there are more than one crystal covering the same part of the protein sequence but they only differ in the fact that they have been crystallized with different ligands or cofactors all of them will be considered and will be shown in the list of results. If the same protein is crystallized in complex with another protein it will be considered separately and it will appear with the information of the partner protein. As we check online the UniProt database to obtain the list of all the crystals, each job will show the results available at the time of your submission.As an example, UniProt accession P01112 (Human HRAS), has more than one hundred crystals, but most of them are covering the same part of the sequence. When HRAS is requested our protocol analyzes only five crystals. Here we are showing three of them: two with the same coverage of the sequence, but interfacing with different proteins and another in a tetramer:
Binding residuesEach structure of the Protein Data Bank provides as metadata, for each heteroatom crystallized, the residues that binds to that heteroatom. A data set of solvent molecules were built to no consider the residues interacting with them as binding residues. The information is parsed directly for the pipeline and the corresponding residues are labeled as involved in binding.Also, fpocket sofware is computed over each protein structure considered for the analysis. Those pockets were the Druggability Score provided is greater than 0.5 are considered, and if a ligand is within it, then all the residues belonging to the pocket (even those not in contact with the ligand) are considered as binding residues. The Catalytic Site Atlas (CSA) is a database documenting enzyme active sites and catalytic residues in enzymes. Classification of catalytic residues were defined including only those residues thought to be directly involved in some aspect of the reaction catalysed by an enzyme. CSA contains 2 types of entry: original hand-annotated entries, derived from the primary literature and homology determined entries, found by sequence comparison methods to one of the original entries. The residues belonging to the CSA database are labeled as catalytic residues for the pipeline. Folding variation energyAll the mutations that were mapped within any protein structure are analyzed with the FoldX software. The FoldX software can be used to predict the energetic impact of a mutation on protein stability or on the stability of a protein-protein complex. The energetic variation provoked for each mutation is computed with the BuildModel command, and those mutations with more than 1kcal/Mol energetic impact are labeled as destabilizing mutation in the residue list of the website.For homomeric structures, each mutation's variation energy is computed only in one of the chains, assuming that the impact of that mutation on the other chains will have a similar impact. Residue exposure analysisFor each residue of each structure, the Solvent Accessible Surface Area (SASA) was computed and also the full Area. The percentage of exposure of the sidechain of the residues are informed, and those with more than the 50% exposed are labeled as exposed residue in the website. For this computation, the PyMol sofware is used as an command line tool calling to the get_area feature.The glycine residue always present a 0% of exposure, because of its absence of sidechain. Interface analysisTo label a residue as interface involved, FoldX software were used, searching for those residues belonging to the interface between two chains with a distance criteria, within the specific crystal that it is been analyzed. Also, the database 3did is used to inform if the position that the residue occupies in the protein family were labeled as interfacing to another protein in any crystal of any protein in its family.Determine if a mutated residue could belong to an interface its crucial to predict if it will affect or not its function. Other propertiesIn order to analyze the mobility of each residue, the BFactor annotated in the crystal is informed. This information could be useful because those mutations happening in flexible zones are expected to be less influential than those occurring in rigid zones. A progress bar informs where is the BFactor value located relatively to all the values in the structure. Also the accessibility is informed relatively to the other residues of the structure.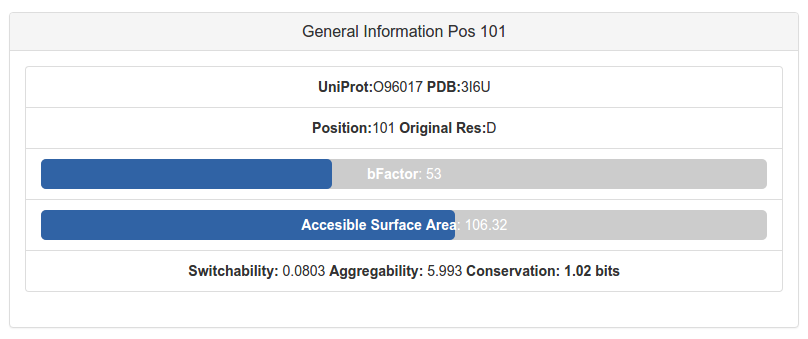
As can be seen, other properties are informed: the Switchability informs the propensity of residues to provoke a switch from alpha helix to beta sheet, which is a destabilizing effect in the protein; the Aggregability is the propensity of a residue to generate aggregation when is muted, and is computed using Tango Software. Finally, the Conservation is the value in bits of the corresponding letter of the original aminoacid in the PFam family if it is assigned: mutate very conserved positions is expected to be influential in the protein folding. |
Tutorials and examples
Tutorials:
Tutorial: Job loading instructions. Tutorial: Explained output. Help: Interpreting results. Sample results: PAH: Phenylalanine-4-hydroxylase NRAS: GTPase NRas HRAS: GTPase HRas KRAS: GTPase KRas BRAF: Serine/threonine-protein kinase B-raf RAF1: proto-oncogene serine/threonine-protein kinase RASA1: Ras GTPase-activating protein 1 NF1: Neurofibromin MAP2K1: Dual specificity mitogen-activated protein kinase kinase 1 MAP2K2: Dual specificity mitogen-activated protein kinase kinase 2 PTPN11: Tyrosine-protein phosphatase non-receptor type 11 SOS1: Son of sevenless homolog 1 SPRED1: Sprouty-related, EVH1 domain-containing protein 1 |